We study African sleeping sickness.
African sleeping sickness, or human African trypanosomiasis, is a devastating human disease affecting sub-Saharan Africa. It is fatal if left untreated, and existing drugs are extremely toxic. Sleeping sickness, along with nagana, an animal disease caused by the same parasite, costs sub-Saharan Africa billions of dollars per year. The causative agent of both human and animal trypanosomiasis is the protozoan parasite Trypanosoma brucei.

A hypothetical model of IgM antibody (teal) binding to VSG (pink and blue).

Antigenic variation leads to waves of parasitemia as each variant is recognized.
Antigenic variation allows T. brucei to escape immune recognition.
T. brucei is covered by a dense variant surface glycoprotein, or VSG, coat. The highly antigenic VSG is so densely packed on the parasite cell surface that it effectively obscures all other antigens from immune recognition (left, top). Under normal circumstances, this would ensure quick recognition and clearance of the parasite, but T. brucei undergoes "antigenic variation": the parasite can change the way it looks to the immune system by turning on a new VSG, using a genomic repertoire of ~2000 different VSG-encoding genes. As the VSG expressed by a population of parasites is recognized by the immune system and then cleared, a minority of parasites will “switch” their VSG coat. These “switchers” will then be recognized by the immune system, but not before another set of parasites will have turned on another VSG and escaped immune clearance. This pattern results in periodic waves of parasitemia characteristic of T. brucei infection (left, bottom).
The dynamics of antigenic variation are complex.
Antigenic variation is typically depicted as shown above: a single or limited number of VSGs are expressed at any given time, suggesting that infection proceeds through the ordered expression of individual variants. Recent work (Hall et al., 2013; Mugnier et al., 2015) has shown, however, that T. brucei parasites in the bloodstream express a huge diversity of VSGs at any given time during an infection, more like the picture on the right. What's more, another recent study (Trindade & Rijo-Ferreira, 2016) has shown that most T. brucei parasites exist outside of the bloodstream in infected mammals. This represents another niche, totally unexplored (so far!), for even greater VSG diversity.

A more accurate cartoon.

Image adapted from Taylor and Rudenko, 2006.
VSG diversification is probably critical to sustaining an infection.
The diversity of VSGs expressed at any given time suggests that the parasite's genome-encoded VSG repertoire is insufficient to maintain the sometimes years-long infections observed in the wild. We know that parasites can form new "mosaic" VSGs (left) through recombination during infection. It is likely that this kind of VSG diversification extends the genomic VSG repertoire, allowing T. brucei to maintain a long-term infection.
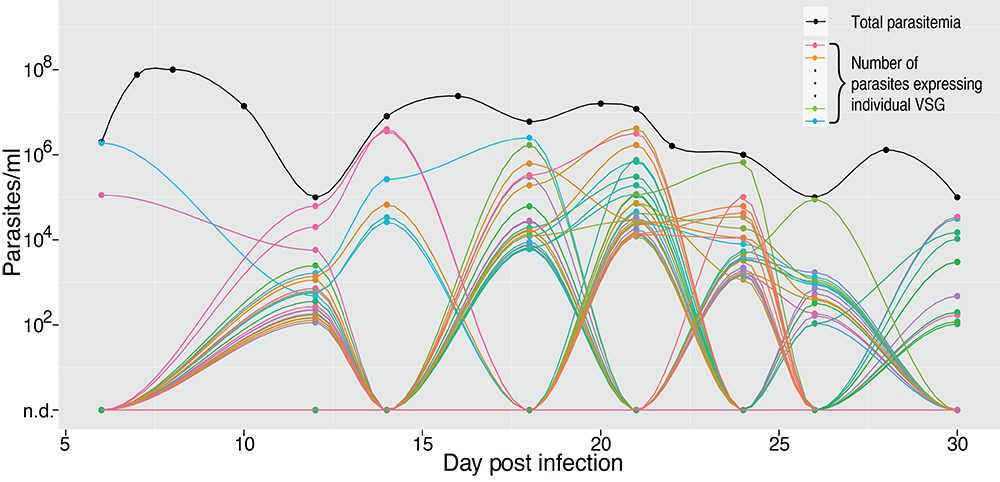
VSG expression, as measured by VSG-seq, in a population during a mouse infection with T. brucei. From Mugnier, 2015.
High-throughput sequencing is an awesome tool for studying antigenic variation.
Historically, resolution has been a limitation for studies of antigenic variation at the population level. Using high-throughput sequencing approaches such as VSG-seq,a technique we developed, we can now measure VSG expression in high resolution. This has led to a better understanding of VSG diversity and expression dynamics in vivo. Long term, we hope it will inform other aspects of antigenic variation as well.
We are are interested in how T. brucei diversifies its VSG repertoire and how this affects infection dynamics.
With all of this in mind, our lab hopes to use high-throughput approaches like VSG-seq to understand how T. brucei creates new VSGs and how this affects the dynamics of infection and antigenic variation. Here are some specific questions we are interested in at the moment:
What determines VSG switching order?
What role do extravascular parasites play in antigenic variation and immune evasion?
How do mosaic VSGs form, and how often?
Can point mutations in VSGs play a role in parasite immune evasion?